



Pubblichiamo un testo a firma del dottor Vincenzo Nigro, membro della CMS UILDM, che approfondisce alcuni degli argomenti trattati durante il webinar "Comprendere la genetica delle malattie neuromuscolari" del 27 ottobre 2020.
Le foto a cui si fa riferimento all'interno del testo sono pubblicate, in sequenza, nello slideshow sopra il testo. Per visualizzarle clicca sopra.
Tutta la sequenza del DNA umano, il genoma, è immensa e fatta di oltre sei miliardi di lettere. Metà è di provenienza paterna e l’altra metà di provenienza materna. Il DNA è localizzato nel nucleo di ciascuna cellula ed è identico tra una cellula e l’altra. Infatti, tutto il genoma deriva dalla ricopiatura fedele del DNA contenuto nel nucleo della prima cellula (zigote). Quando dalla prima cellula per divisioni consecutive si passa a milioni e miliardi di cellule, si assiste ad una progressiva specializzazione: le singole cellule faranno mestieri diversi, rendendosi utili, ad esempio, nella retina, nel muscolo o nel fegato. Questa specializzazione non dipenderà da differenze nel genoma, ma dall’accensione o spegnimento di parti diverse del DNA.
Il genoma può essere paragonato al programma operativo di un computer. Il contenuto è come se fosse all’interno di una coppia di hard disk: uno proveniente dal padre e uno proveniente dalla madre. Come detto, in ogni nostra cellula ci sono virtualmente questi due hard disk.
Vedi figura 1
Il genoma materno e paterno danno già alla prima cellula tutte le istruzioni per la fabbricazione, l’uso e la manutenzione del corpo umano. Le istruzioni sono così raffinate che dallo zigote parte lo sviluppo di un essere umano fatto di centinaia di miliardi di cellule specializzate e coordinate che per 70 o 100 anni ci faranno crescere, sentire, amare, muovere, mangiare, pensare, morire.
Il DNA però è soggetto ad alcuni errori di ricopiatura che per fortuna sono pochissimi e vengono in gran parte corretti. Poi ci sono errori dovuti all’invecchiamento delle cellule, ai danni da mutageni ambientali, a spostamenti di elementi mobili ed infine all’invasione di virus di cui non ci accorgiamo. Se gli errori nel DNA sono confinati alle cellule somatiche, cioè alle cellule di tutto il corpo (cervello, rene, muscolo, fegato, occhio, ecc.), questi non porteranno mai alla trasmissione di una malattia genetica. Solo gli errori del DNA della linea germinale (spermatozoi ed ovociti) potranno essere trasmessi. Per questo la linea germinale è isolata dal resto e protetta: lì è contenuto il DNA destinato ai figli. Se ci sarà una nuova mutazione nella sola linea germinale il figlio sarà mutato, ma i genitori no. Si parlerà in questo caso di mutazione de novo.
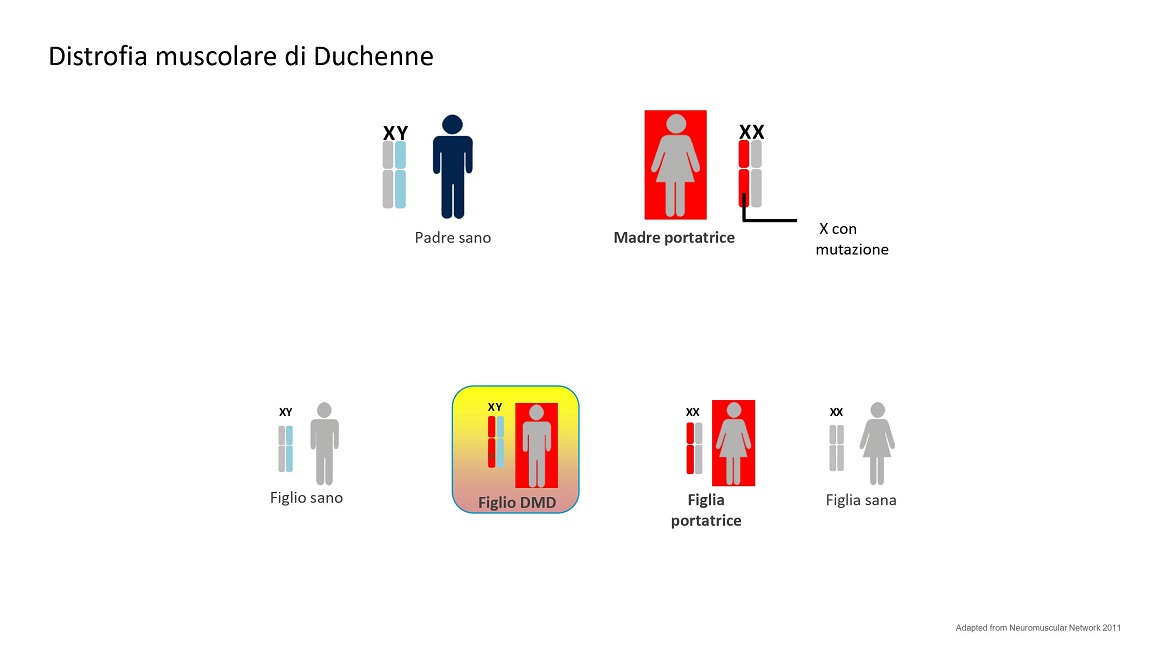
La distrofia muscolare di Duchenne è una malattia importante delle cellule muscolari che agisce progressivamente. Alla nascita la funzione dei muscoli sembra normale e poi anno dopo anno peggiora. Per riconoscere precocemente questa malattia è di fondamentale importanza la misurazione di un enzima del siero, conosciuto come CK o creatina chinasi, che aumenta moltissimo in seguito ad un danno muscolare. La distrofia di Duchenne è causata dalle mutazioni all’interno di un gene lunghissimo localizzato sul cromosoma X e particolarmente esposto ad alcuni meccanismi di mutazione, quali le grandi delezioni o duplicazioni. A queste si aggiungono le consuete piccole mutazioni di singole lettere del DNA (mutazioni puntiformi) che colpiscono ogni parte del genoma.
In linea teorica, un bambino Duchenne è un maschio figlio di una madre portatrice da cui ha ricevuto la copia mutata del gene presente su cromosoma sessuale X. Non ha alcun ruolo il padre, perché al figlio maschio trasmette il cromosoma Y. Quindi una donna portatrice ha la probabilità al 50% di trasmettere la malattia, se nasce un maschio, e il 50% di trasmettere lo stato di portatrice se nasce una femmina.
Vedi figura 2
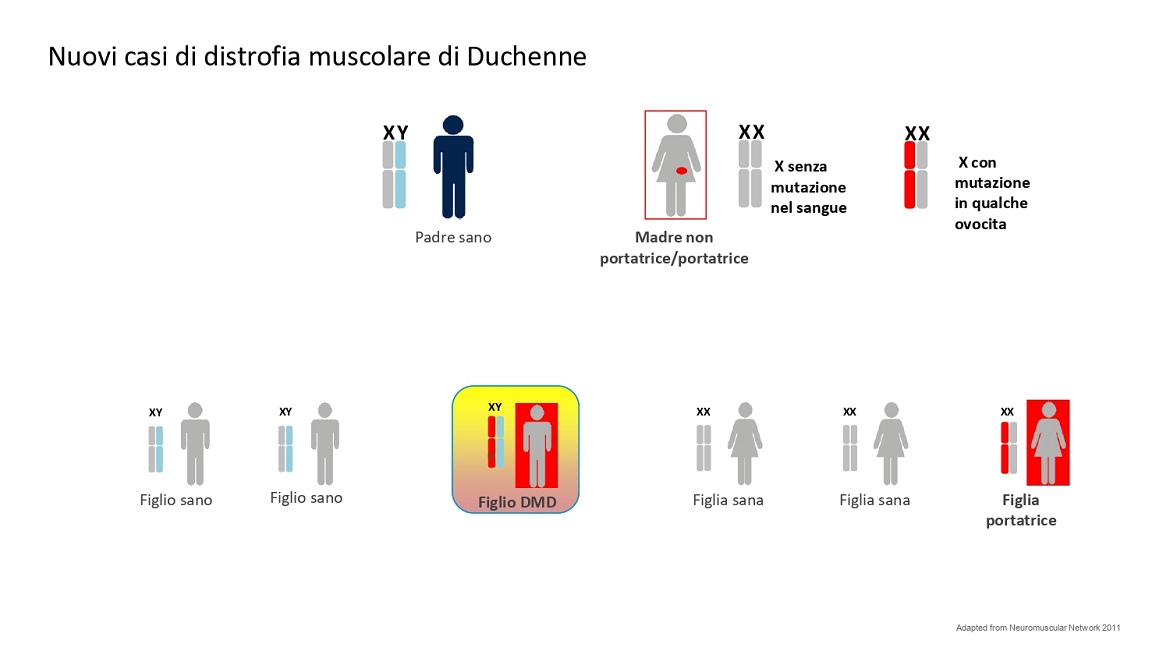
Un bambino Duchenne può anche nascere da una donna apparentemente non portatrice. Se si fa un esame del DNA dal sangue di questa donna questo risulterà normale. In realtà la mutazione potrebbe essere localizzata solo nella linea germinale (mutazione de novo). Questa donna pur non essendo apparentemente portatrice è comunque a rischio di avere altri figli maschi affetti o figlie femmine portatrici.
Vedi figura 3
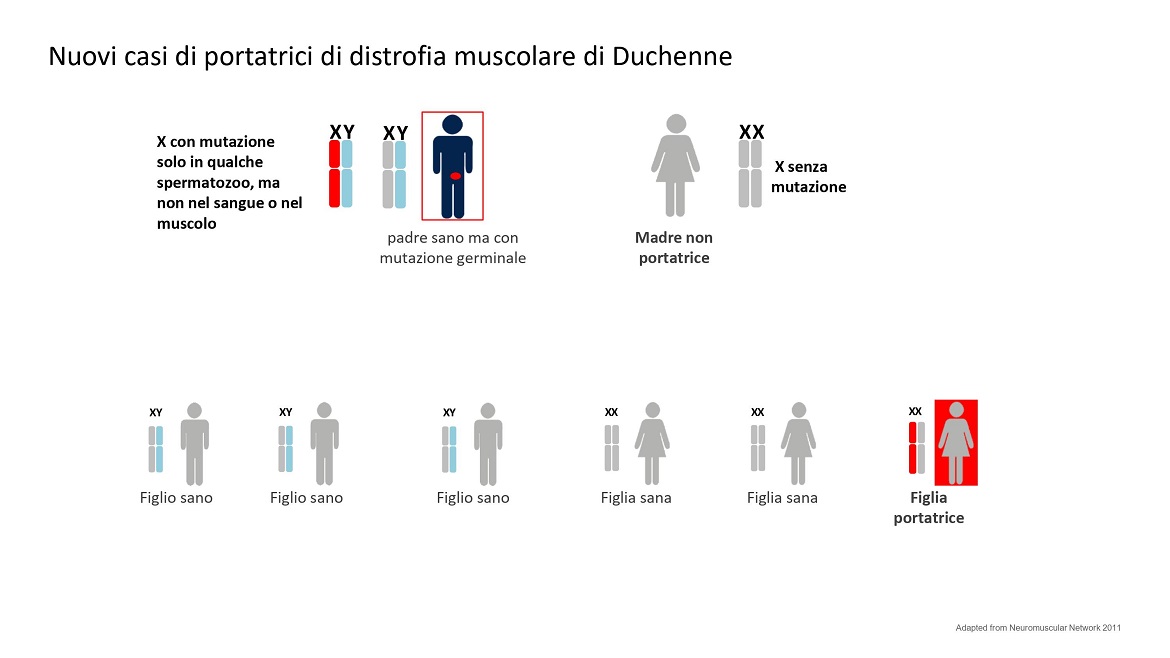
Anche gli spermatozoi possono avere una mutazione de novo. Anzi questa è molto più frequente con l’avanzare dell’età del padre. C’è una tendenza ad avere figli più tardi e questo comporta più mutazioni de novo nella linea germinale. La mutazione del Cromosoma X paterno si trasmette solo alle figlie femmine che così diventeranno portatrici mentre non avrà effetto sui figli maschi.
Vedi figura 4
C’è da chiarire la differenza tra delezioni, duplicazioni ed il motivo per cui in alcuni casi la distrofia è di Duchenne ed in altri casi di Becker. L’effetto di ciascuna di queste mutazioni può essere più o meno grave in rapporto alla possibilità di produrre la proteina distrofina. Se l’informazione genetica è sfalsata da una delezione, nessuna distrofina può essere prodotta e si ha la distrofia muscolare di Duchenne. Nel caso in cui si può ancora produrre una distrofina, sia pure priva di una porzione centrale, si ha una distrofia muscolare meno grave, nota come Becker.
Se l’informazione genetica è sfalsata da una delezione o duplicazione, potrebbe essere rimessa in cornice da un’ulteriore delezione. Come succede quando l’ascensore si blocca tra due piani e un altro piccolo spostamento la riporta al piano. La rimessa in cornice è stata tentata con i tanti oligonucleotidi antisenso che sono stati sperimentati sinora per produrre il “salto di alcuni esoni”.
Le mutazioni puntiformi invece possono contenere un segnale di stop prematuro (mutazione nonsenso) o far sfalsare l’informazione genetica o far perdere solo alcuni segmenti. L’effetto di ciascuna di queste mutazioni può essere più o meno grave in rapporto alla possibilità di produrre la proteina distrofina.
Le mutazioni nonsenso sono speciali perché il gene è praticamente intatto, ma è sbagliato un segnale. Su questi segnali sbagliati si interviene con l’Ataluren un farmaco che forza la lettura di questo segnale sbagliato (cosiddetto “readthrough”). Ovviamente lo stesso farmaco non può avere effetto in altri casi perché non c’è un segnale sbagliato, ma una strada mancante.
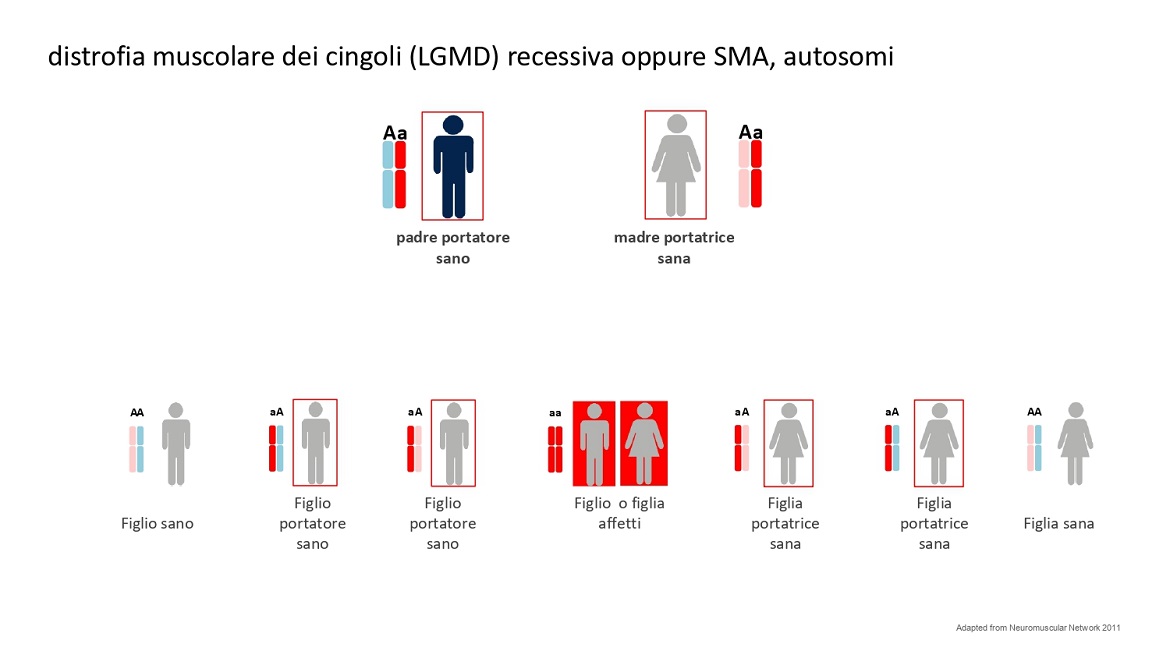
Un meccanismo di trasmissione ereditaria totalmente differente c’è nelle distrofie muscolari dei cingoli (dette LGMD) e nella atrofia muscolare spinale (SMA).
Il meccanismo di trasmissione ereditaria si dice autosomica recessiva. Questo significa semplicemente che i portatori sono entrambi i genitori e che il bambino per essere malato deve avere una doppia mutazione. Nell’esempio degli hard disk deve avere un difetto comune nello stesso file. Ovviamente meno portatori sani ci sono e meno è probabile che due portatori uguali si incontrino. In questo tipo di trasmissione ereditaria le nuove mutazioni sono rare.
Vedi figura 5
Purtroppo, i portatori sani di SMA solo in Italia sono oltre un milione e ci sono molti nuovi casi di SMA estremamente gravi in famiglie in cui non c’era un precedente. Il gene responsabile si chiama SMN1 e quando è mutato si ha un danno nei neuroni spinali che comandano i movimenti dei muscoli. Si dà il caso che esista un gene simile, detto SMN2 ma poco attivo. Oggi esistono delle terapie che mirano a riaccendere SMN2 ed i risultati sono molto buoni se il trattamento è precoce. Di qui la necessità di fare la diagnosi di SMA il prima possibile e quindi questo spiega l’utilità degli screening neonatali.
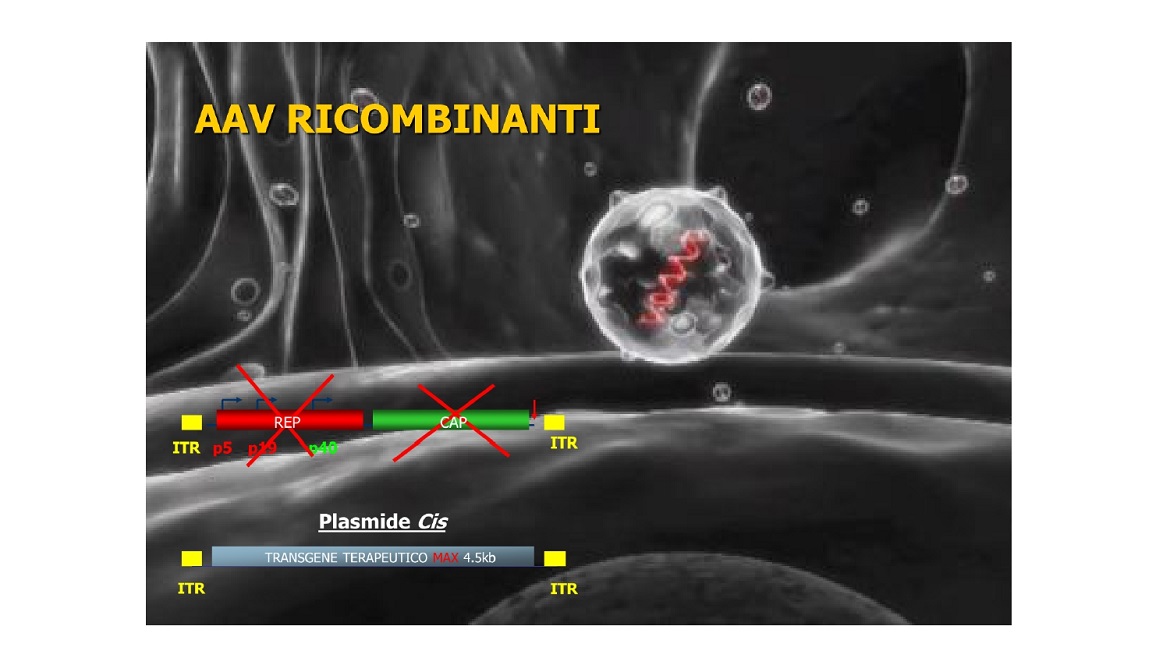
L’ultima frontiera della terapia mirata è l’uso dei virus adeno-associati noti come AAV e la loro importanza strategica per la terapia genica. Questi virus sono innocui e spesso ci infettano e restano nell’organismo a lungo senza generare una risposta immunitaria. Questo ha convinto i ricercatori a tentare di usare gli AAV per correggere un difetto genetico causato dall’assenza di attività di un gene. Gli AAV agiscono solo come trasportatori delle copie intatte di un gene. Nessun gene del virus è lasciato al suo posto, ma l’AAV stesso è prima svuotato e resta solo un involucro proteico di trasporto. Questa terapia sta avendo grande successo in tante malattie genetiche a partire dalla correzione dei difetti della retina. Anche la SMA può essere trattata e a breve anche le LGMD, in quanto il trasportatore ha un limite di carico per i geni troppo grandi.
Vedi figura 6
Purtroppo, il gene della distrofia muscolare di Duchenne è molto grande, mentre quelli delle LGMD sono più piccoli. Ma anche per la DMD si sta tentando una strategia con la cosiddetta “microdistrofina”, una versione “tipo Becker”. Questi tentativi potrebbero a breve portare degli ottimi risultati.
Poi ci sono malattie rarissime per cui è necessario leggere l’intero genoma. Cosa si vede quando leggiamo il genoma? Innanzitutto ci accorgiamo che siamo tutti molto simili, ma anche diversi, 4 milioni di lettere differenti tra ciascuno di noi.
Vedi figura 7
Avendo una visione d’insieme possiamo capire che alcune lettere sono più determinanti di altre. Bambini con gravi malattie nuove che non si sanno comprendere e a cui non si trovano nemmeno dei nomi, oggi hanno una diagnosi. Spesso si parla di nuove malattie genetiche e del loro aumento, poi si scopre che i cambiamenti del DNA sono più numerosi nei figli quanti più anni ha il padre.
Leggere il DNA può servire a capire la causa di una malattia, ma anche a comprendere a cosa possa servire ciascuna lettera, guardando cosa succede se quella lettera cambia. Gli errori, o meglio i tentativi della natura, sono quelli che ci hanno fatto evolvere e che oggi ci aiutano a comprendere il significato di quello che è scritto.
È come avere un grande quadro elettrico e per tentativi, abbassando le levette vedere la corrispondenza con le luci che si spengono. Leggere il DNA di sempre più persone ci sta facendo capire sempre meglio quali sono i nostri circuiti e cosa accade se si guastano. È questo il senso del Progetto Telethon Malattie senza diagnosi. Infine, una conclusione significativa: i nostri progressi scientifici e terapeutici sono più veloci in rapporto agli investimenti economici. Avremmo potuto curare molte più malattie solo se avessimo investito di più in passato.
